This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200627) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 9254340. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/bulkRNA-gs_enrichment.Rmd) and HTML (docs/bulkRNA-gs_enrichment.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
This document presents the Gene Set Analysis for the bulk RNA-seq data in this project.
The analyisis is performed using fGSEA (Sergushichev A (2016). “An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation.” bioRxiv. doi: 10.1101/060012, http://biorxiv.org/content/early/2016/06/20/060012).
We sort the genes by their Wald statistic, as provided by DESeq2.
run_fgsea <-function(my_res, pathways) {
my_res <-my_res[complete.cases(my_res),]
# Sort by the statistic (Wald statistic is the default in DESeq2)
my_res <-my_res %>%arrange(stat)
stat <-my_res$stat
names(stat) <-my_res$symbol
# Rn fGSEA
fgseaRes <-fgsea(pathways, stat, nperm=10000)
# Change the e! stable IDs into gene symbols in the leading edge list (col #8)print(htmltools::tagList(
datatable(fgseaRes,
options =list(dom ='frtip')
) %>%formatSignif(columns=c('pval', 'padj', 'ES', 'NES'), digits=3) %>%formatStyle(
'padj',
target ="row",
fontWeight =styleInterval(0.05, c('bold', 'normal'))),
htmltools::tags$br()
))
fgseaRes_sig <-fgseaRes[fgseaRes$padj <0.05,]
fgseaRes_sig <-fgseaRes_sig[order(fgseaRes_sig$NES),]
cat("\n\n")
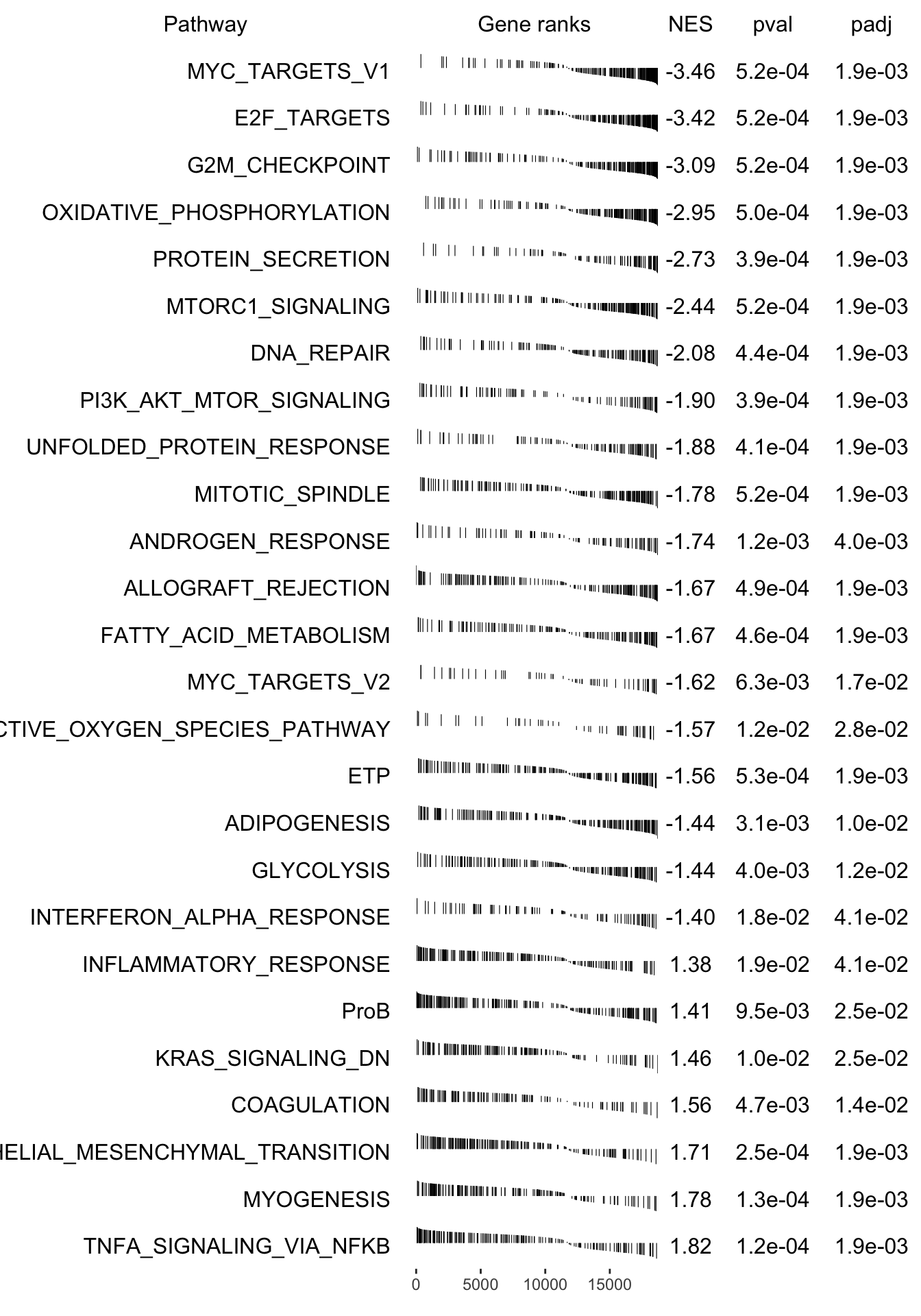
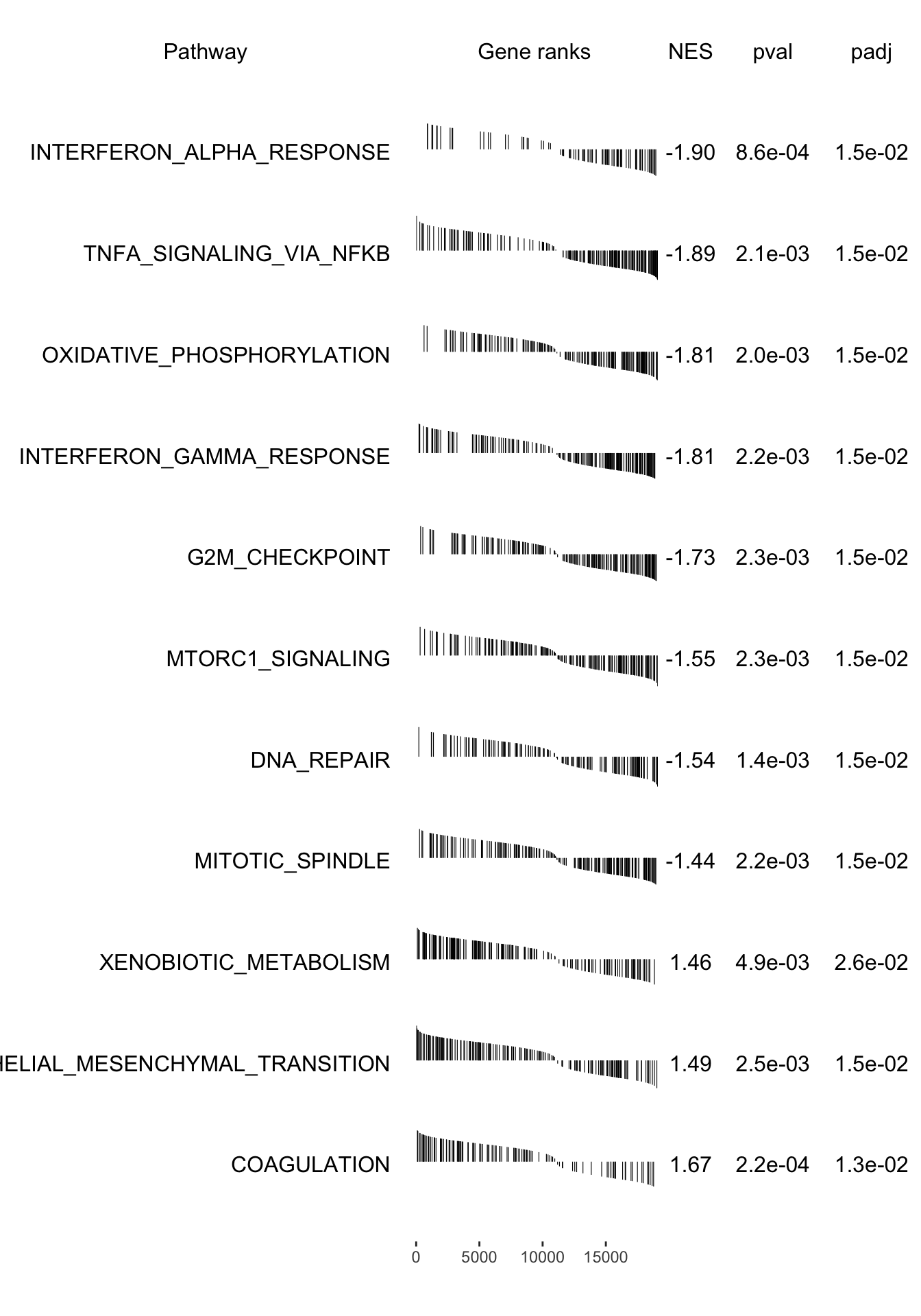
grid::grid.newpage()
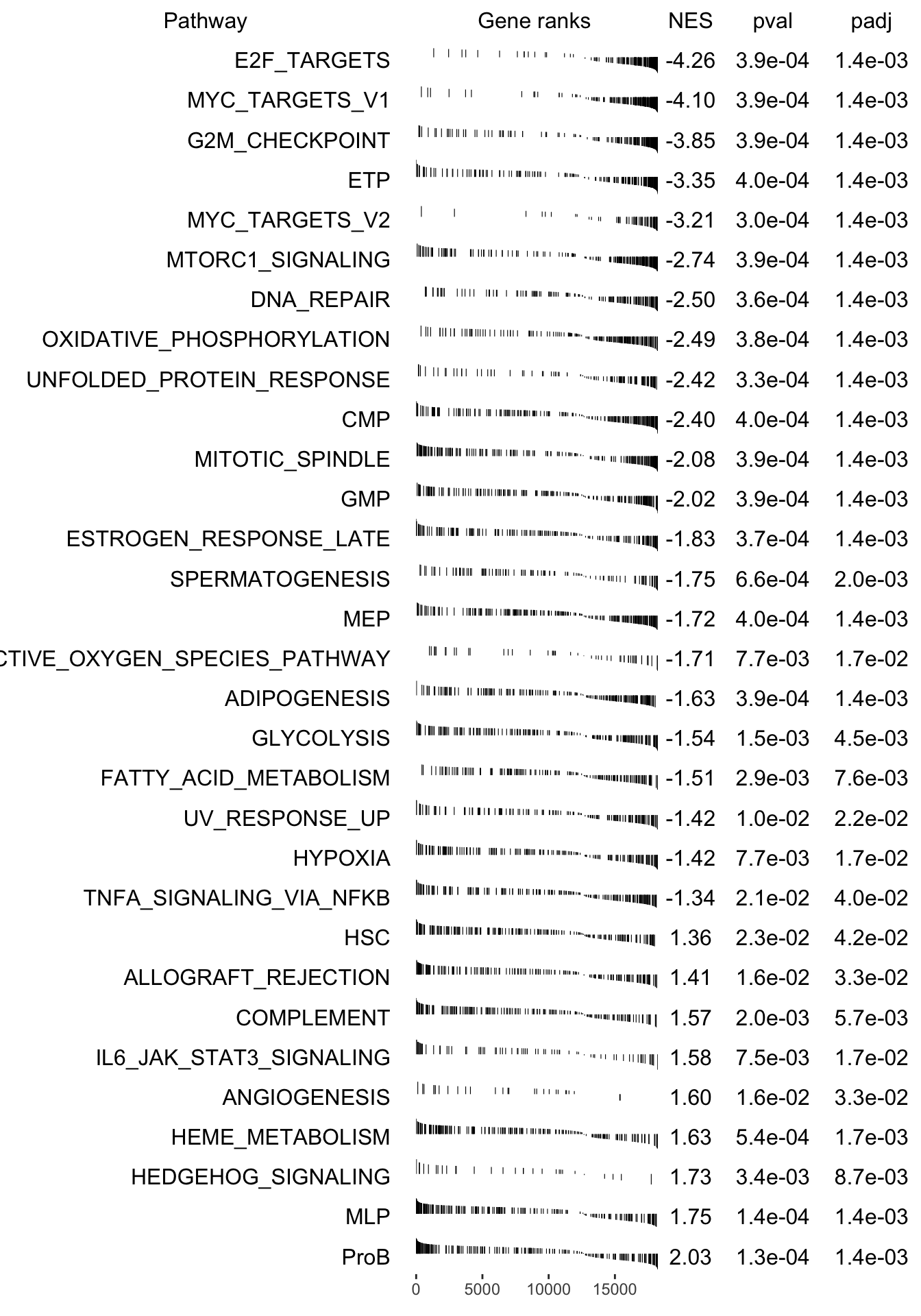
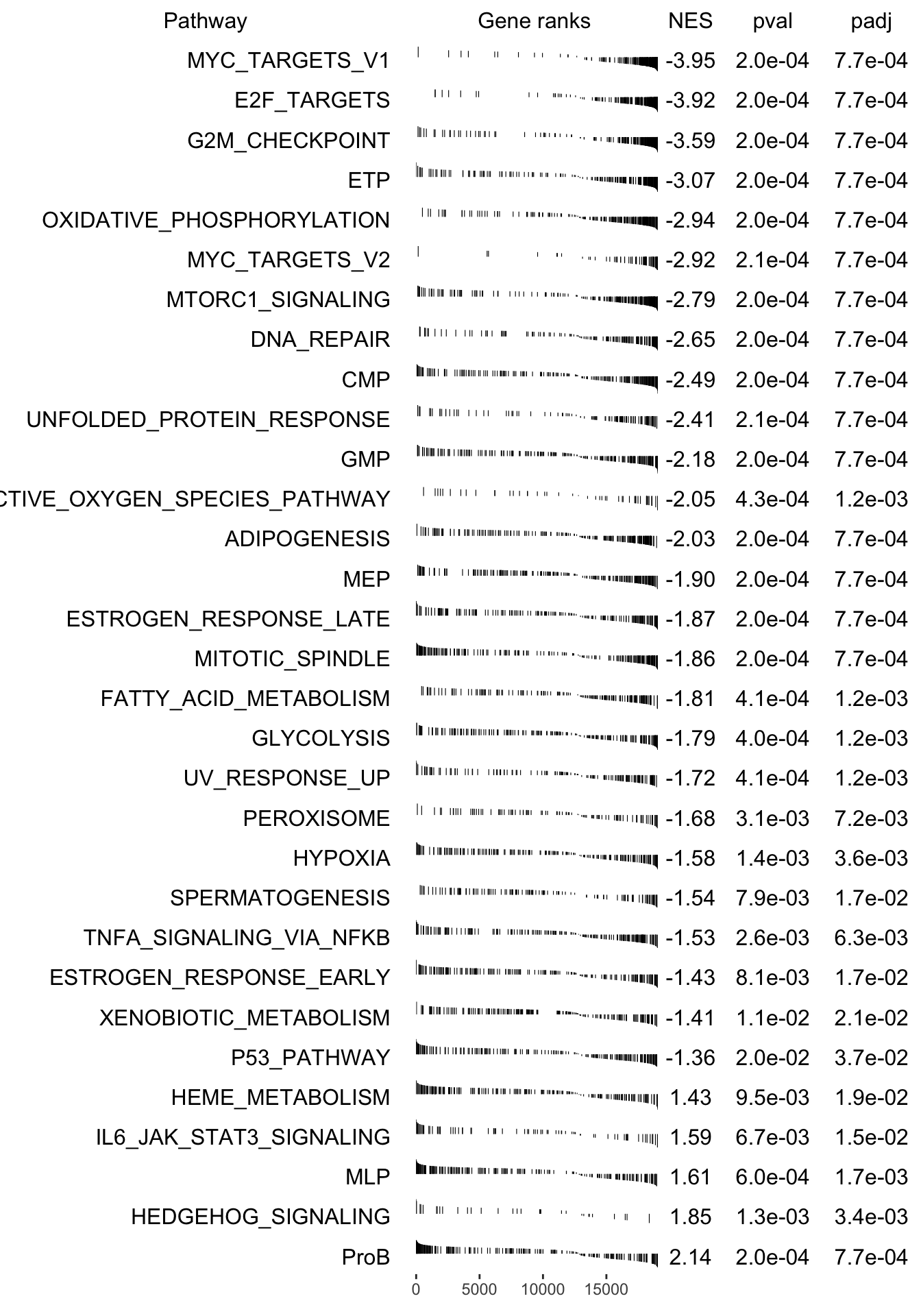
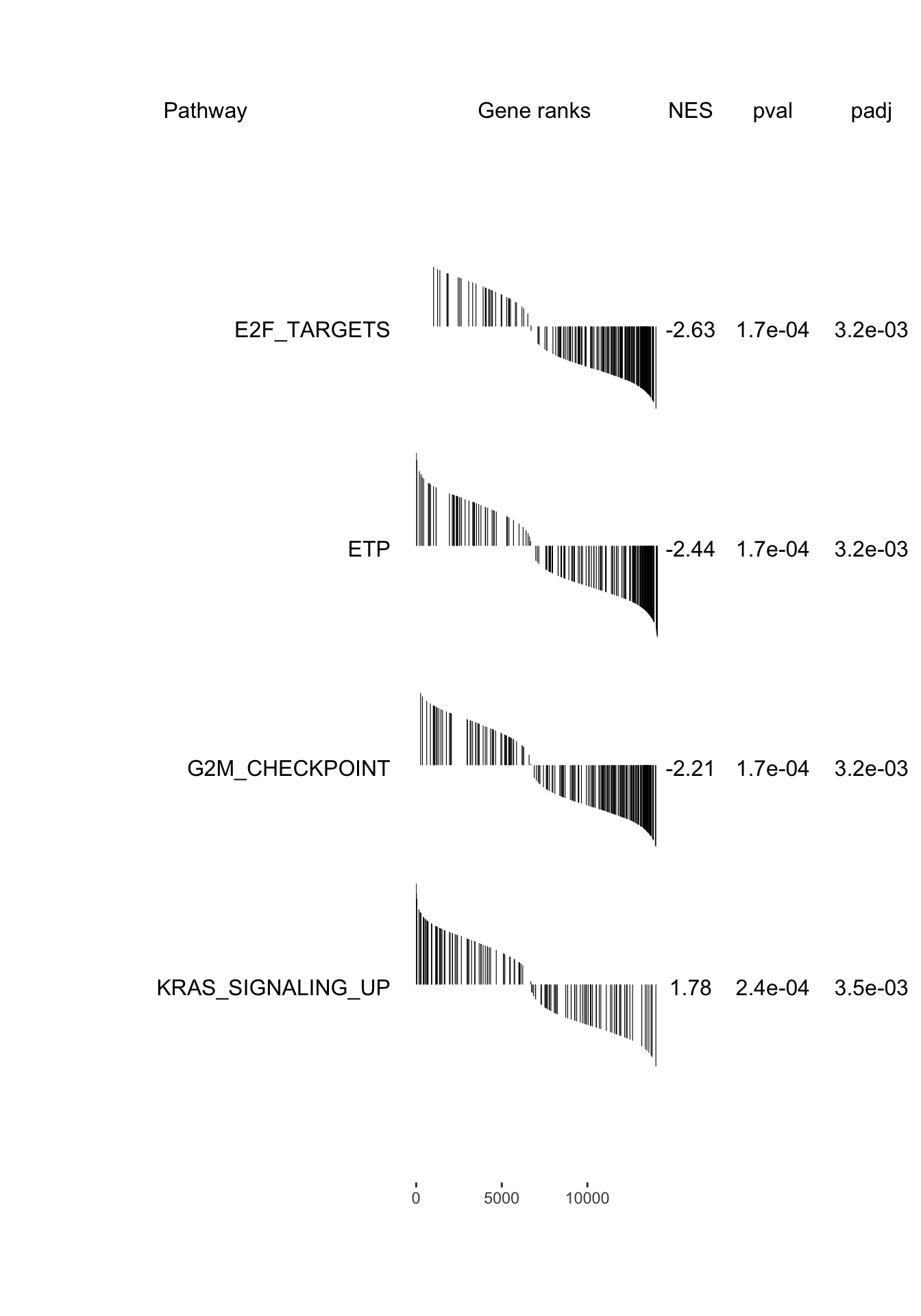
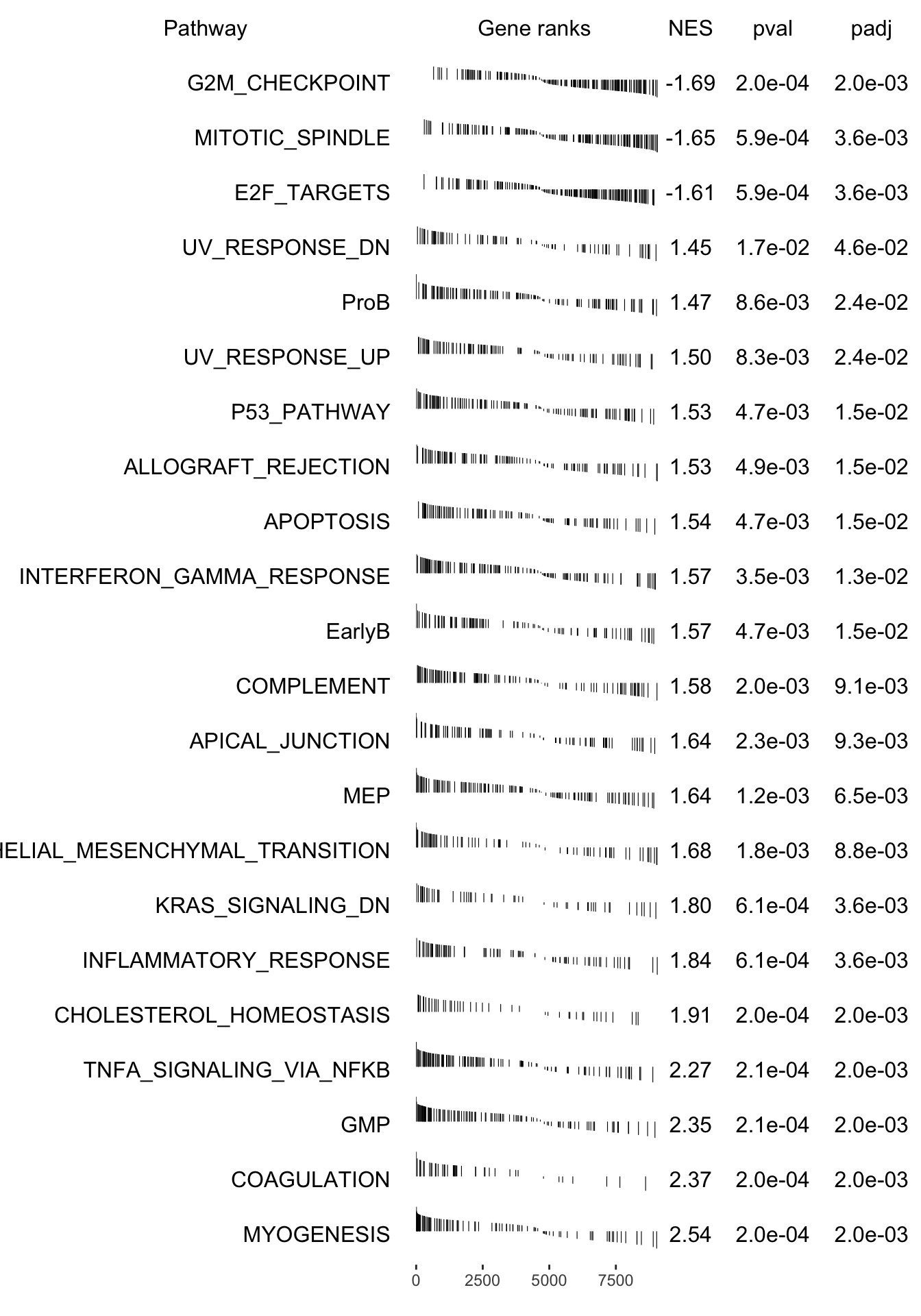
plotGseaTable(pathways[fgseaRes_sig$pathway], stat, fgseaRes_sig, gseaParam =0.4)
cat("\n\n")
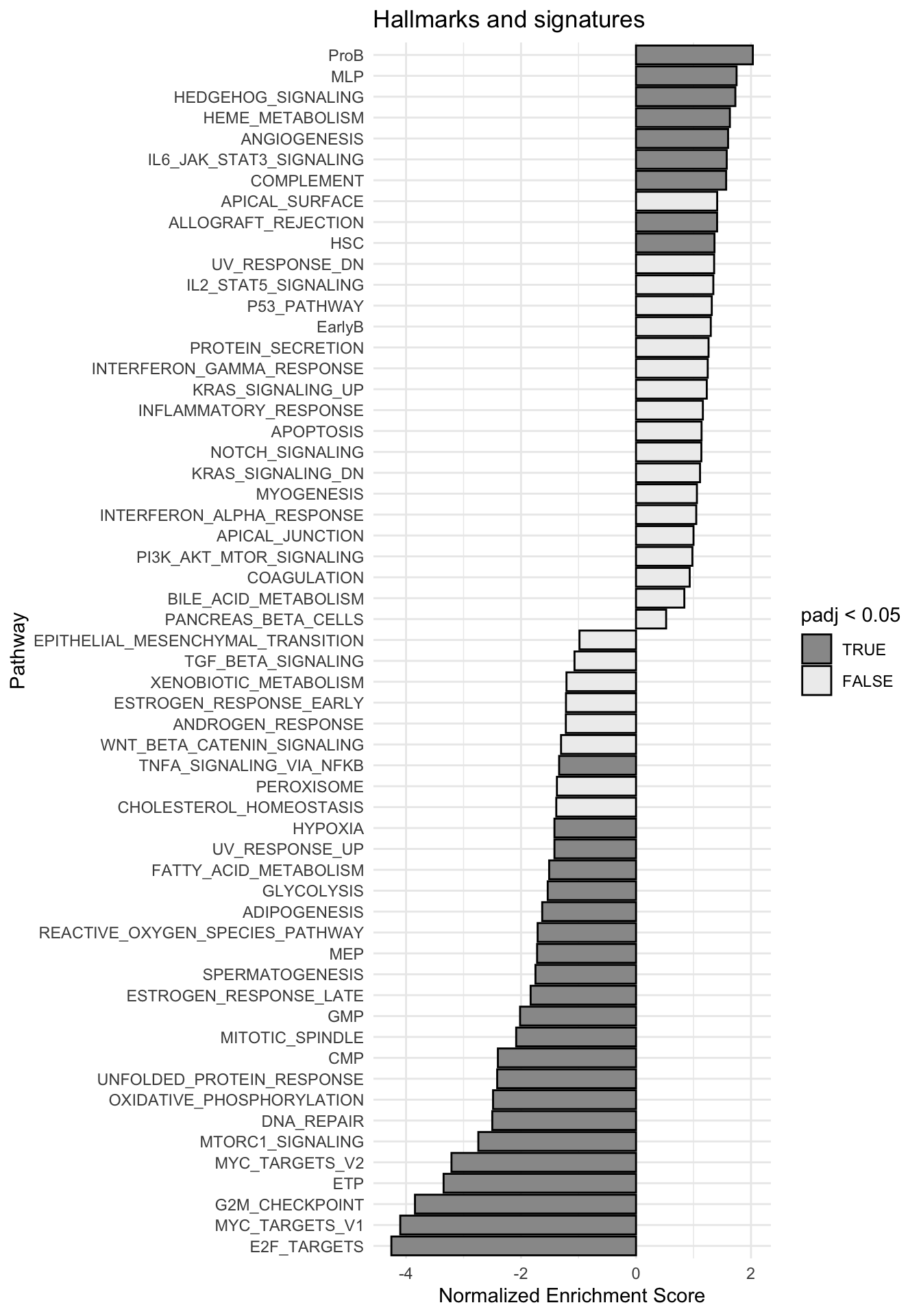
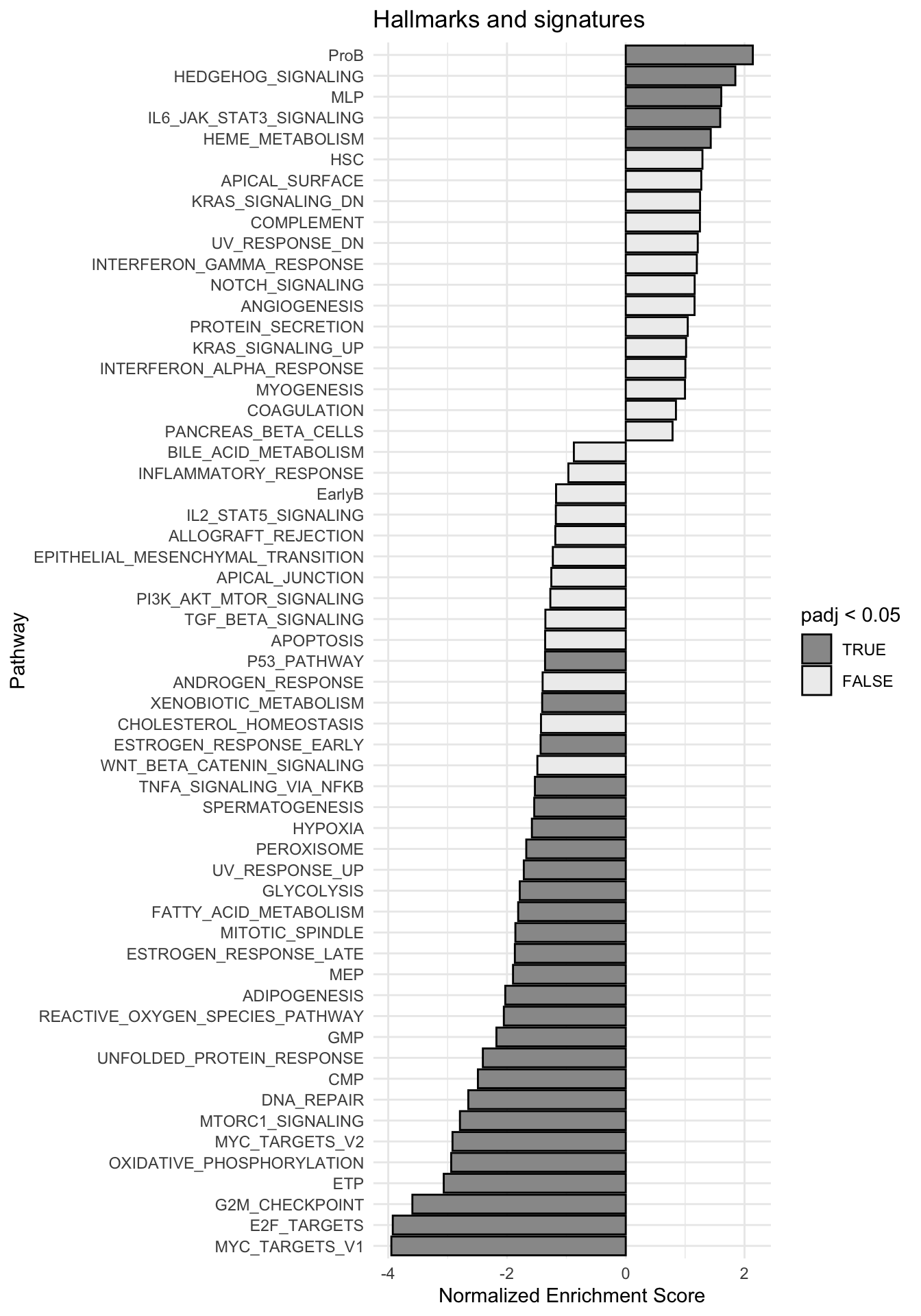
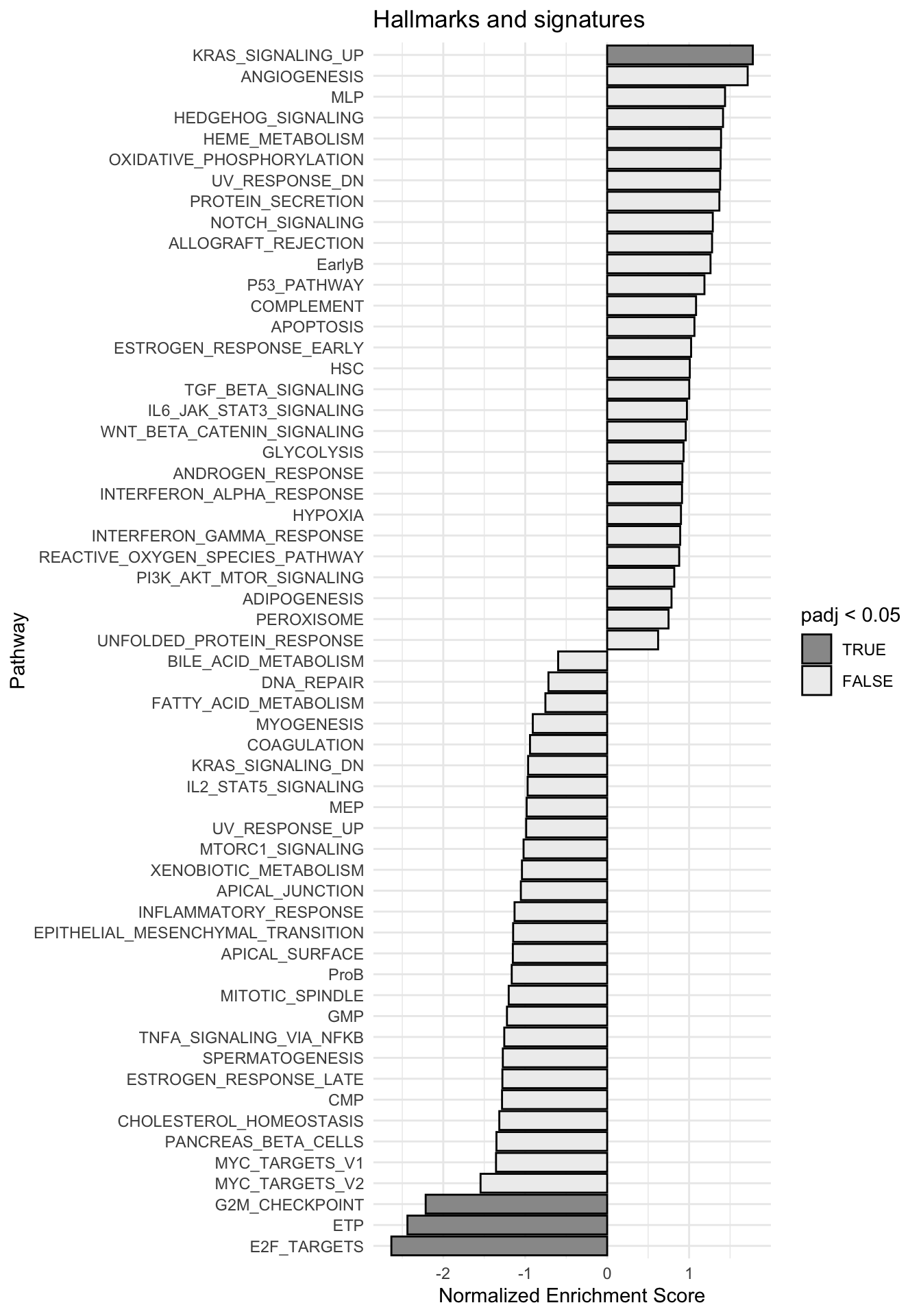
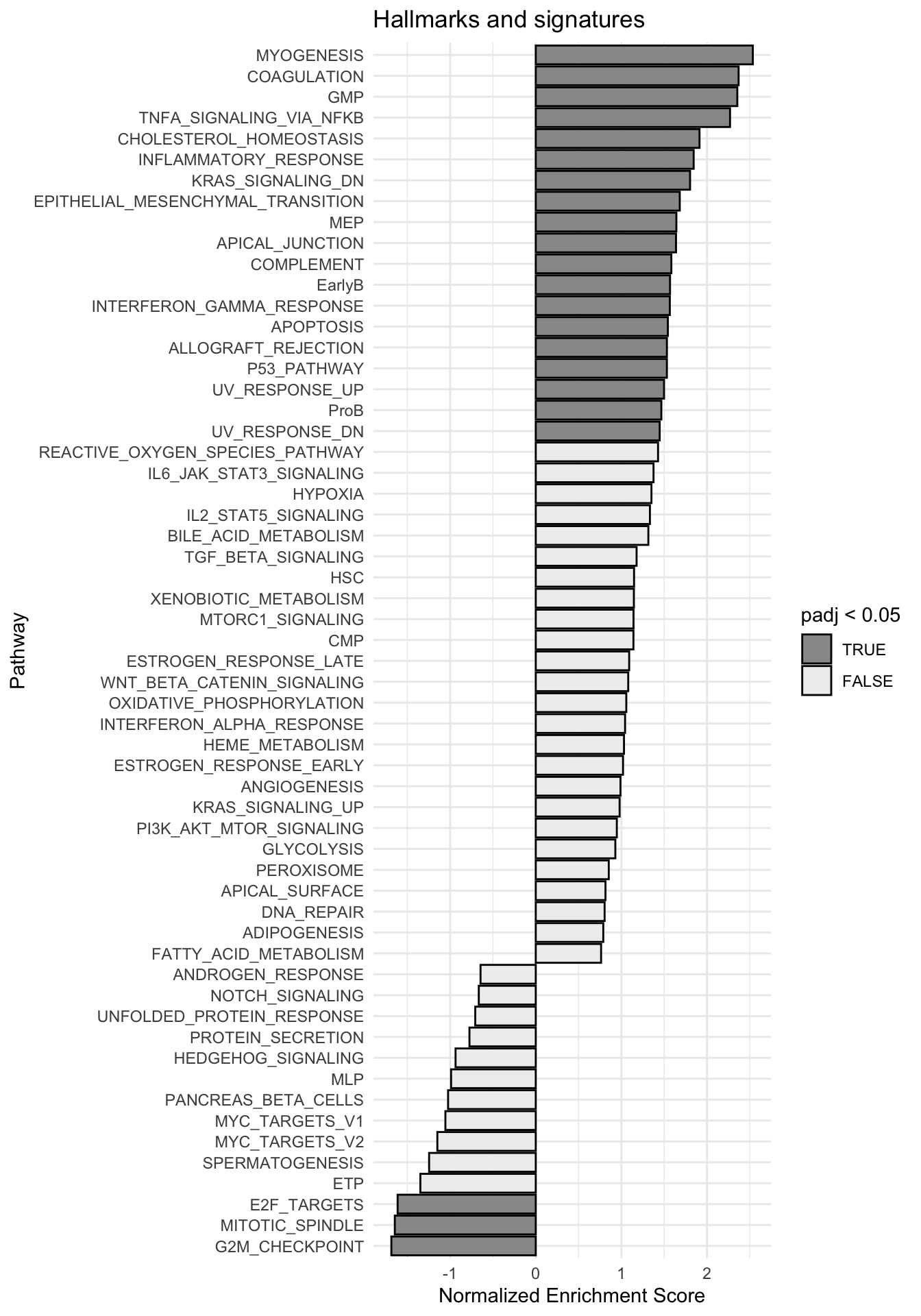
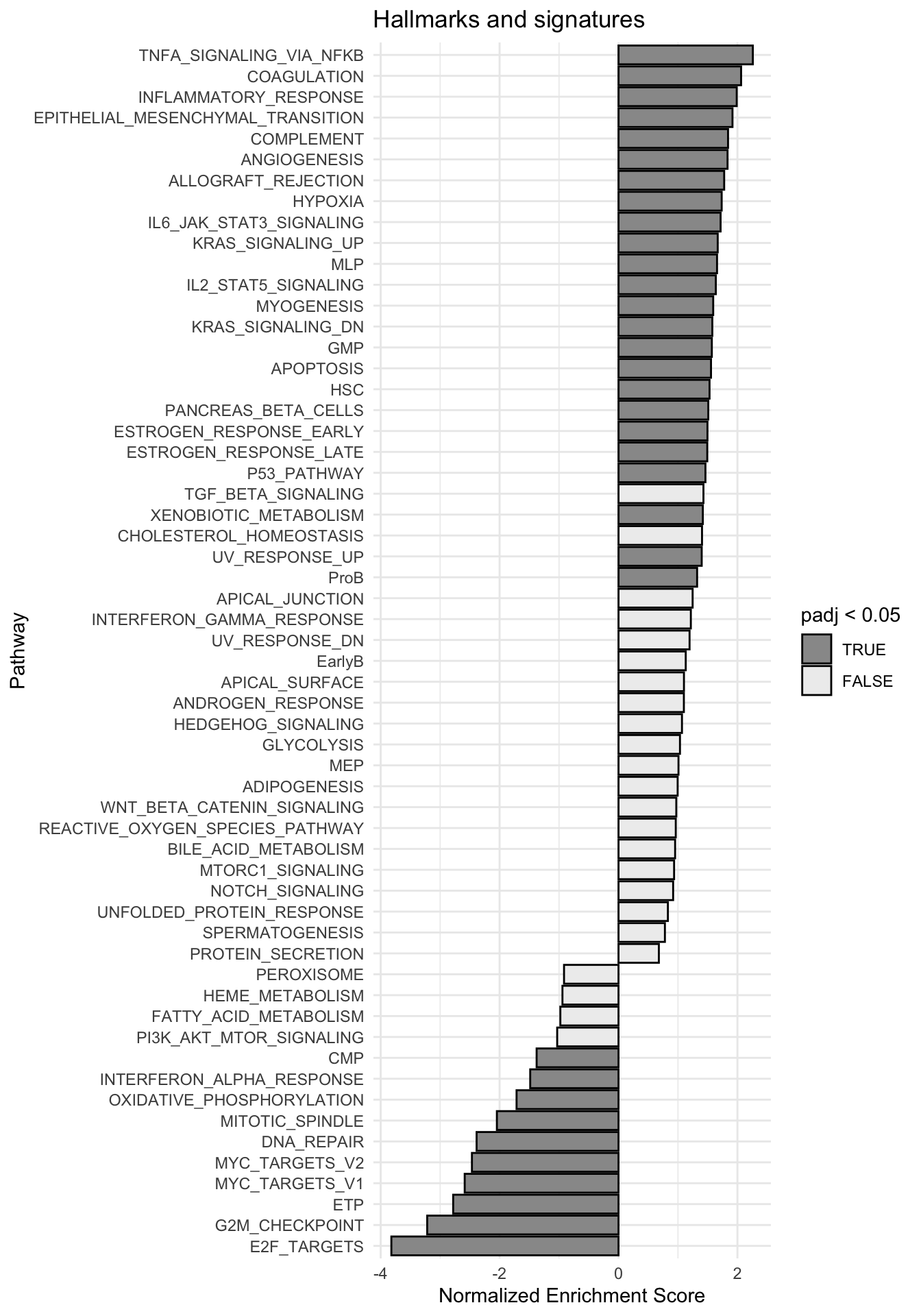
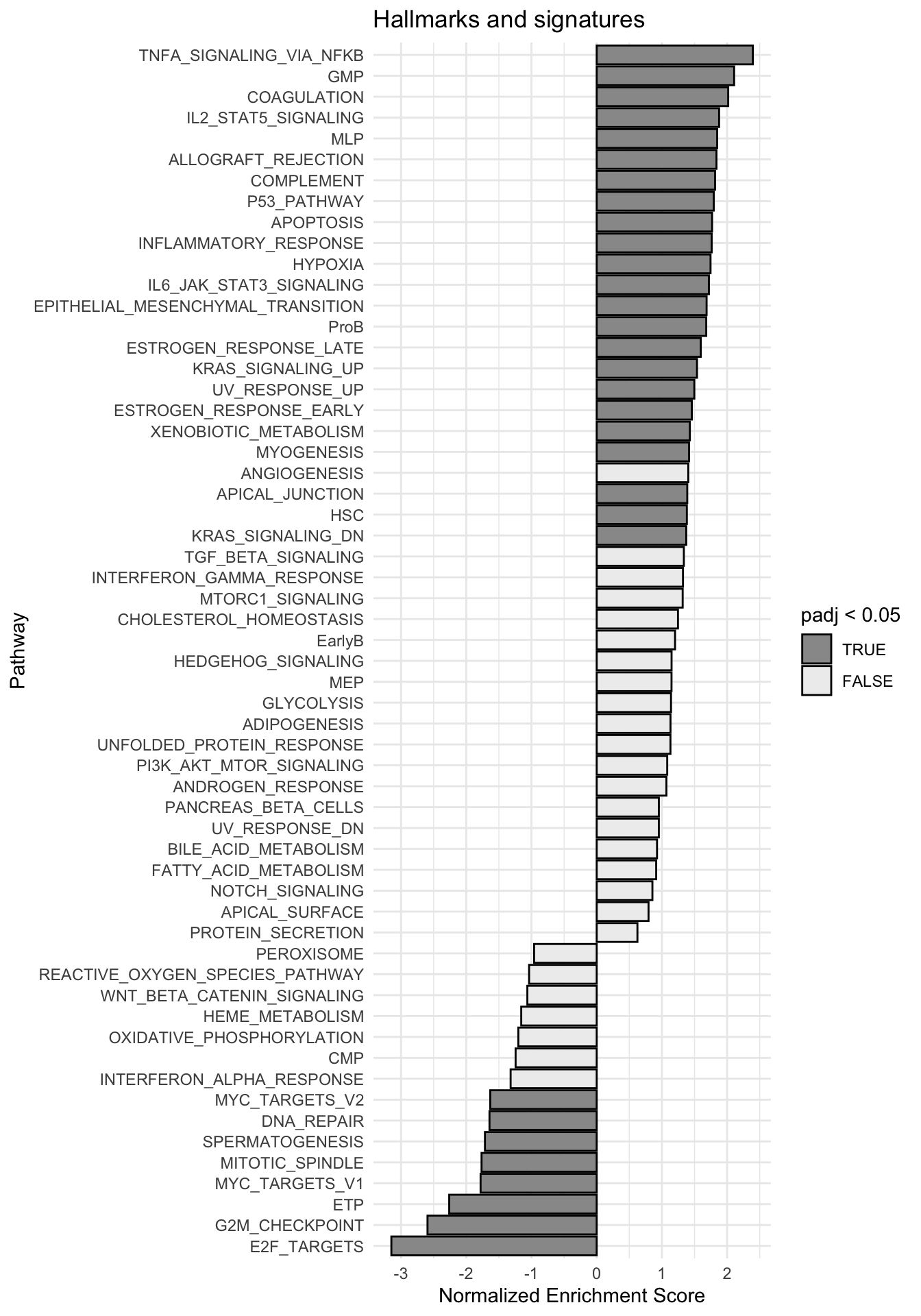
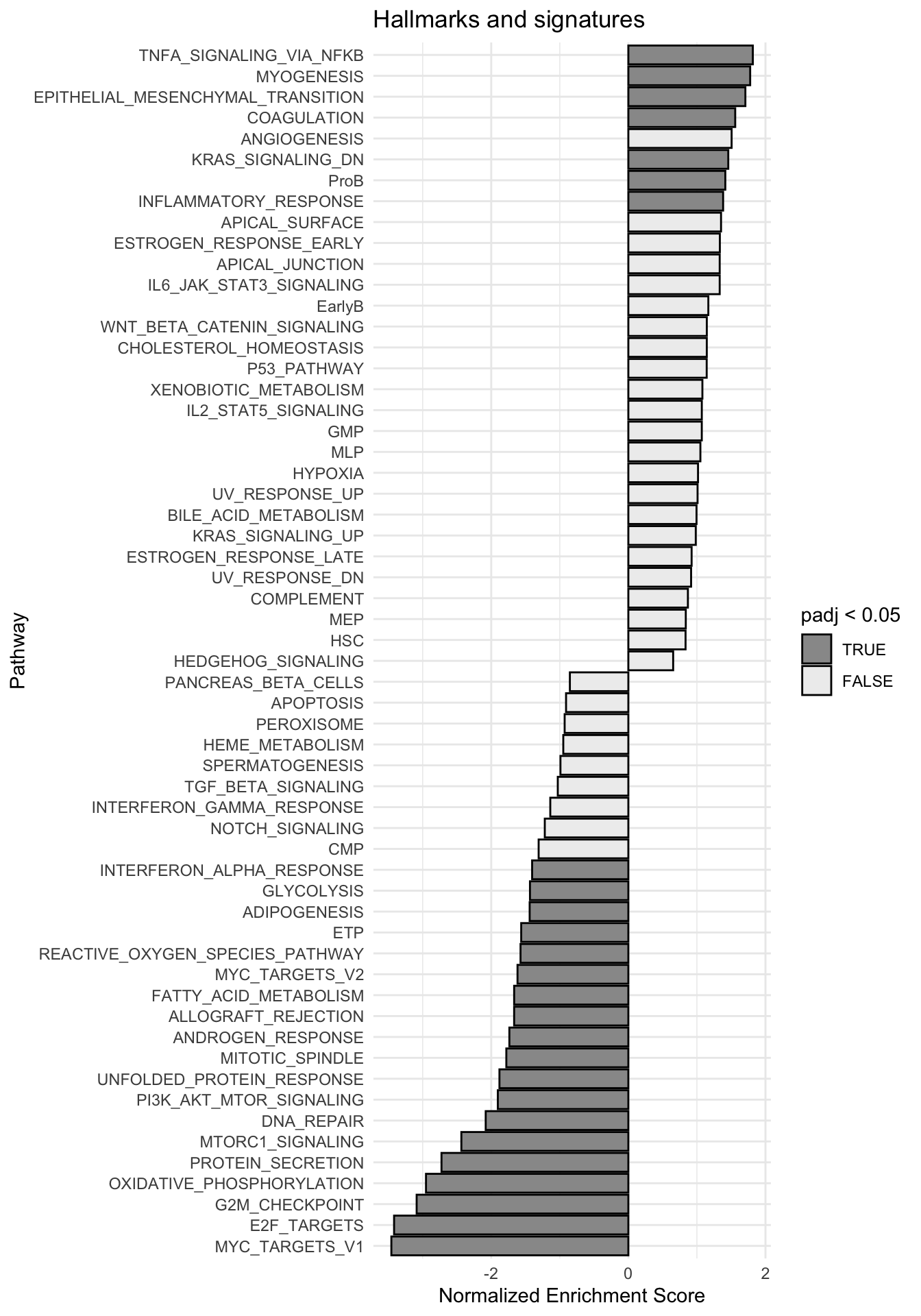
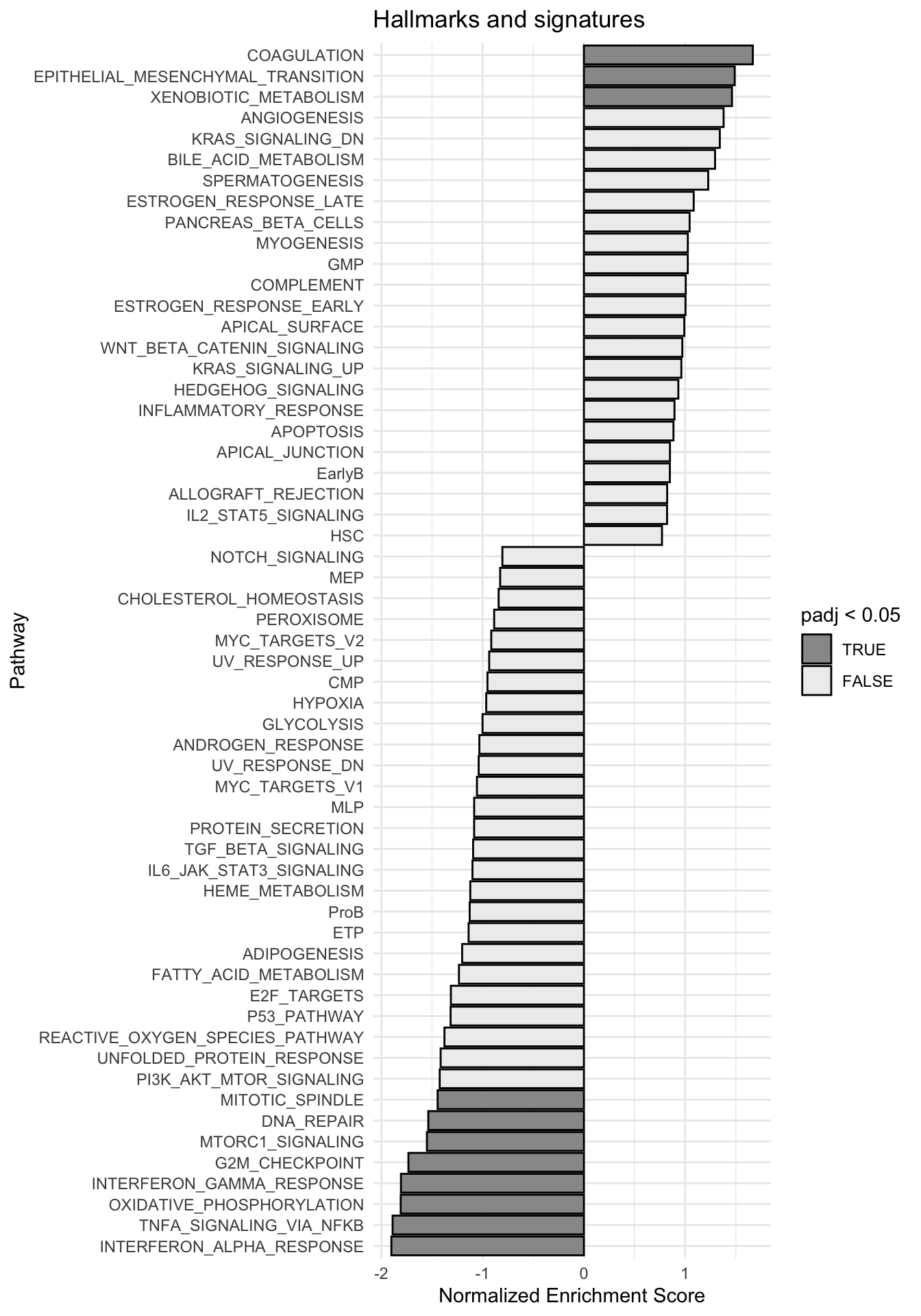
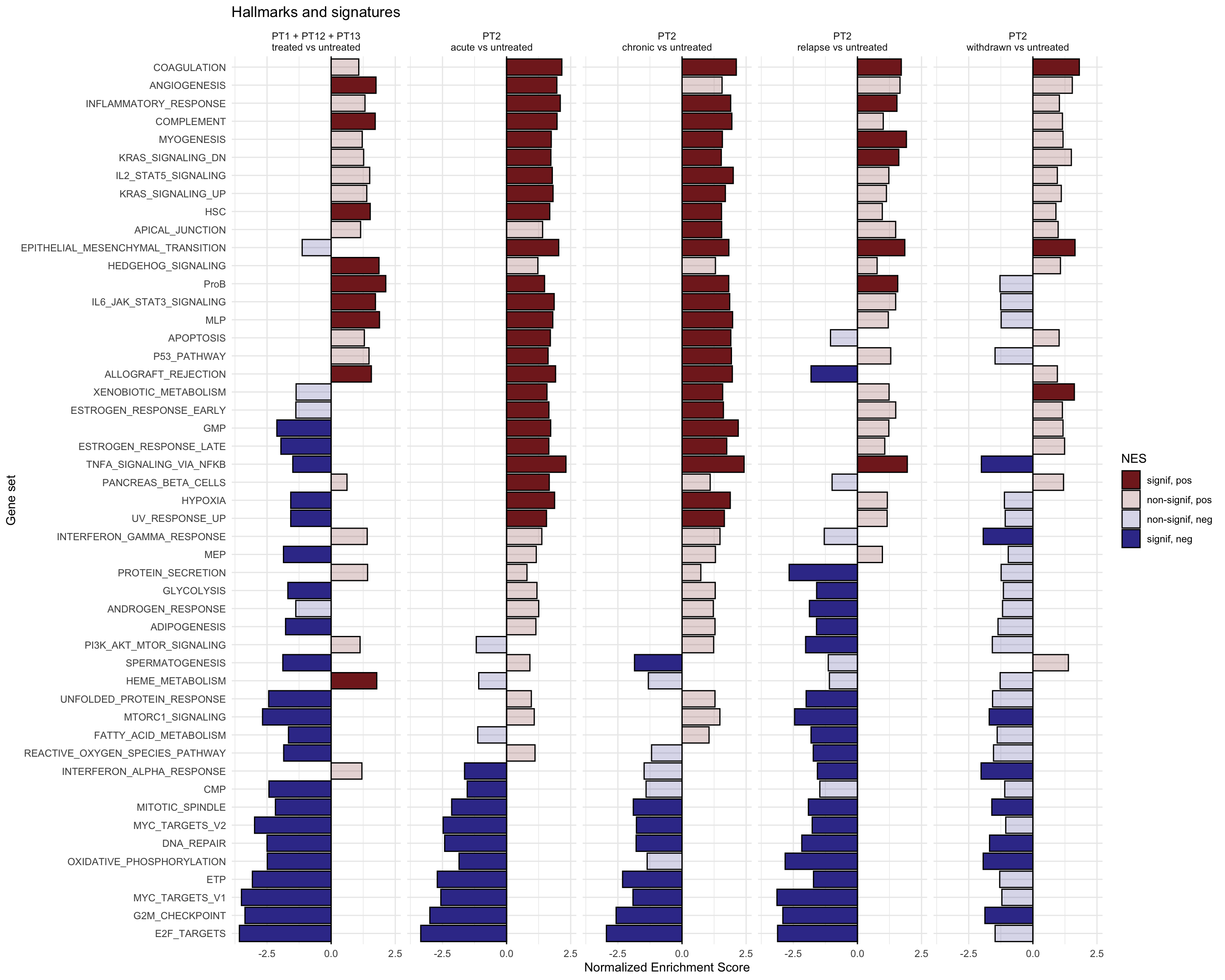
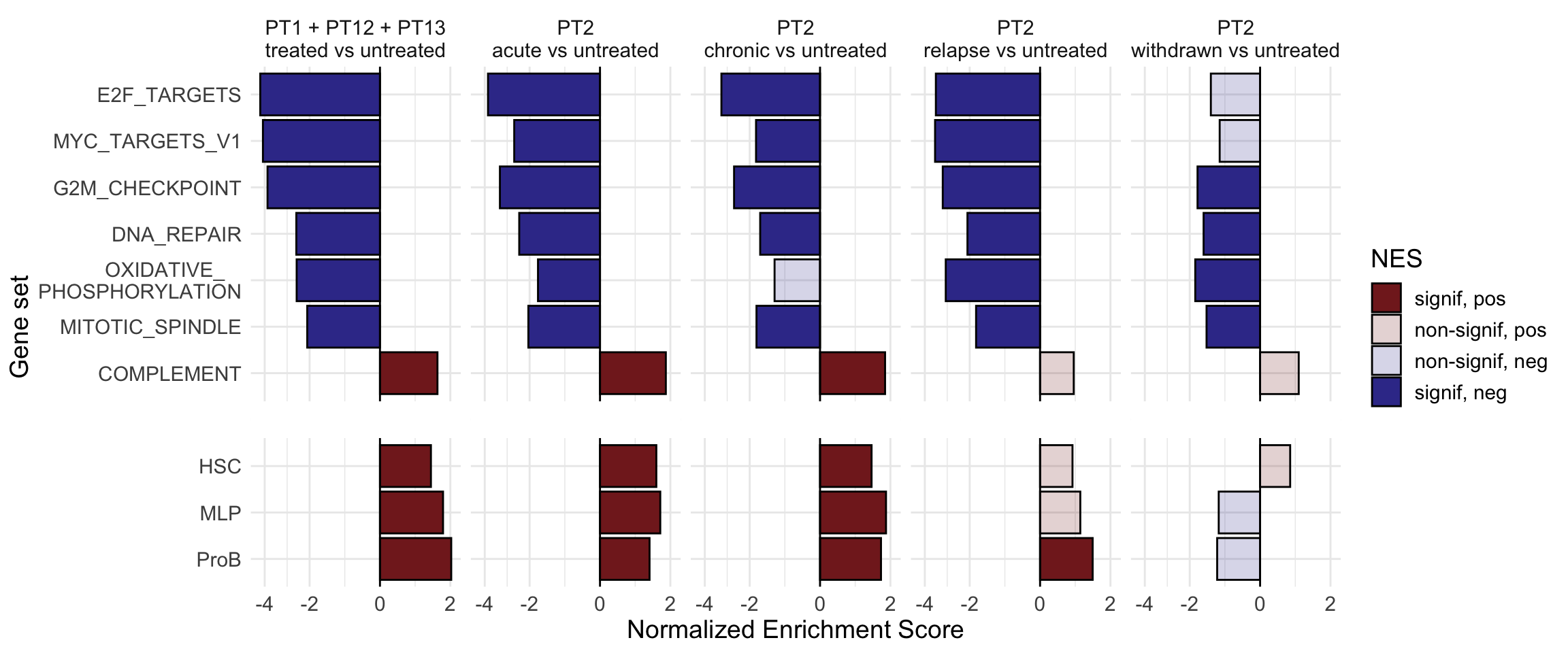
g <-ggplot(fgseaRes, aes(reorder(pathway, NES), NES)) +geom_col(aes(fill=padj<0.05), col ="black") +coord_flip() +labs(x="Pathway", y="Normalized Enrichment Score",
title="Hallmarks and signatures") +scale_fill_manual(limits =c(T, F), values =c("#999999", "#EEEEEE")) +theme_minimal()
print(g)
cat("\n\n")
return(invisible(fgseaRes))
}
Results
fgsea_results <-list()
Treated vs Untreated (PT1 + PT12 + PT13)
bulk_res <-readRDS("output/deseq2-mini_bulk4_dds.3pts-Treated-vs-Untreated.rds")
tag <- "treated_vs_untreated.3pts"
fgsea_results[[tag]] <-run_fgsea(bulk_res, pathways)
Warning in fgsea(pathways, stat, nperm = 10000): There are ties in the preranked stats (0.83% of the list).
The order of those tied genes will be arbitrary, which may produce unexpected results.
Warning in fgsea(pathways, stat, nperm = 10000): There are duplicate gene names,
fgsea may produce unexpected results
bulk_res <-readRDS("output/deseq2-mini_bulk4_dds.pt1-Treated-vs-Untreated.rds")
tag <- "treated_vs_untreated.pt1"
fgsea_results[[tag]] <-run_fgsea(bulk_res, pathways)
Warning in fgsea(pathways, stat, nperm = 10000): There are ties in the preranked stats (0.84% of the list).
The order of those tied genes will be arbitrary, which may produce unexpected results.
Warning in fgsea(pathways, stat, nperm = 10000): There are duplicate gene names,
fgsea may produce unexpected results
bulk_res <-readRDS("output/deseq2-mini_bulk4_dds.pt12-Treated-vs-Untreated.rds")
tag <- "treated_vs_untreated.pt12"
fgsea_results[[tag]] <-run_fgsea(bulk_res, pathways)
Warning in fgsea(pathways, stat, nperm = 10000): There are ties in the preranked stats (0.86% of the list).
The order of those tied genes will be arbitrary, which may produce unexpected results.
Warning in fgsea(pathways, stat, nperm = 10000): There are duplicate gene names,
fgsea may produce unexpected results
bulk_res <-readRDS("output/deseq2-mini_bulk4_dds.pt13-Treated-vs-Untreated.rds")
tag <- "treated_vs_untreated.pt13"
fgsea_results[[tag]] <-run_fgsea(bulk_res, pathways)
Warning in fgsea(pathways, stat, nperm = 10000): There are ties in the preranked stats (1.29% of the list).
The order of those tied genes will be arbitrary, which may produce unexpected results.
Warning in fgsea(pathways, stat, nperm = 10000): There are duplicate gene names,
fgsea may produce unexpected results
bulk_res <-readRDS("output/deseq2-mini_bulk4_dds.pt2-Acutely treated-vs-Never treated.rds")
tag <- "acute_vs_never"
fgsea_results[[tag]] <-run_fgsea(bulk_res, pathways)
Warning in fgsea(pathways, stat, nperm = 10000): There are ties in the preranked stats (0.84% of the list).
The order of those tied genes will be arbitrary, which may produce unexpected results.
Warning in fgsea(pathways, stat, nperm = 10000): There are duplicate gene names,
fgsea may produce unexpected results
bulk_res <-readRDS("output/deseq2-mini_bulk4_dds.pt2-Chronically treated-vs-Never treated.rds")
tag <- "chronic_vs_never"
fgsea_results[[tag]] <-run_fgsea(bulk_res, pathways)
Warning in fgsea(pathways, stat, nperm = 10000): There are ties in the preranked stats (0.84% of the list).
The order of those tied genes will be arbitrary, which may produce unexpected results.
Warning in fgsea(pathways, stat, nperm = 10000): There are duplicate gene names,
fgsea may produce unexpected results
bulk_res <-readRDS("output/deseq2-mini_bulk4_dds.pt2-Relapse-vs-Never treated.rds")
tag <- "relapse_vs_never"
fgsea_results[[tag]] <-run_fgsea(bulk_res, pathways)
Warning in fgsea(pathways, stat, nperm = 10000): There are ties in the preranked stats (0.84% of the list).
The order of those tied genes will be arbitrary, which may produce unexpected results.
Warning in fgsea(pathways, stat, nperm = 10000): There are duplicate gene names,
fgsea may produce unexpected results
bulk_res <-readRDS("output/deseq2-mini_bulk4_dds.pt2-Treatment withdrawn-vs-Never treated.rds")
tag <- "withdrawn_vs_never"
fgsea_results[[tag]] <-run_fgsea(bulk_res, pathways)
Warning in fgsea(pathways, stat, nperm = 10000): There are ties in the preranked stats (0.83% of the list).
The order of those tied genes will be arbitrary, which may produce unexpected results.
Warning in fgsea(pathways, stat, nperm = 10000): There are duplicate gene names,
fgsea may produce unexpected results